JDFTx恒电势计算方法与应用
简要介绍: JDFTx是一种平面波密度泛函理论(DFT)代码,旨在易于开发和使用。 JDFTx是使用高度模板化和面向对象的C++11代码编写的,目的是在DFT++代数框架中表达所有物理特性,同时保持较小的内存占用并支持一系列硬件架构(如使用CUDA的GPU),而不需要为每种架构手动优化实现。
相比于最流行的DFT软件VASP所需要的输入文件:
INCAR # 输入文件 设置计算参数
KPOINTS # K点文件 设置k点
POSCAR # 结构文件 包含晶胞和原子坐标信息
POTCAR # 赝势文件 包含原子种类和赝势信息JDFTx 所有命令的设置都可以只写在一个输入文件command.in,JDFTx 支持把结构文件直接以命令的形式写在输入文件里
输入文件是一个命令列表,每行一个;
命令可以按任何顺序出现;根据你的喜好将它们分组;
#后一行中的所有内容都被视为评论并被忽略;
空格分隔单词;多余的空格将被忽略;
\ 号表示续行。
其中最基本的结构输入格式如下:
command <value>
command <key1> <value1> <key2> <value2> ...
以Si为例的布里渊区采样
硅具有金刚石结构,立方晶格常数为5.43埃(10.263玻尔半径)。每个立方晶胞包含8个硅原子(位于顶点、面心和两个半晶胞体心)。但为了更简洁地描述周期性,我们可以使用更小的面心立方晶胞,仅包含2个硅原子:
# ----------------POSCAR----------------
# 设置晶胞
lattice face-centered Cubic 5.43
latt-scale 1.88973 1.88973 1.88973
# 也可以自己设置晶胞,如下
# lattice \
# 10 0 0 \
# 0 10 0 \
# 0 0 10
coords-type lattice # 根据晶格矢量指定原子坐标(分数坐标)也可以选择笛卡尔坐标
ion Si 0.00 0.00 0.00 0
ion Si 0.25 0.25 0.25 0
# ----------------POSCAR----------------
# ----------------POTCAR----------------
# 指定赝势 JDFTx 将会根据你指定的原子种类自动到赝势文件库里选择
ion-species GBRV/si_pbe.uspp
# 也可以使用下面的命令,这里$ID 是变量,会遍历所有的原子,只需要1行命令即可导入所有的赝势
# ion-species GBRV/$ID_pbe.uspp
# ----------------POTCAR-----------------
# ----------------KPOINTS----------------
# Gamma-centered网格(默认):
kpoint 0 0 0 1.
# Monkhorst-Pack网格:
# kpoint 0.5 0.5 0.5 1.
kpoint-folding 8 8 8 # 设置k点密度 8x8x8
# ----------------KPOINTS----------------
# ----------------INCAR----------------
elec-cutoff 20 100 # 波函数和电荷密度的平面波动能截断
dump-name Si.$VAR # 输出的文件名 其中$VAR是文件后缀变量,这里设置Si.$VAR后,输入原子位置文件.ionpos 将会是Si.ionpos
dump End Ecomponents ElecDensity # 输出能量分量和电子密度
# ----------------INCAR----------------将以上内容保存为Si.in,运行:
jdftx -i Si.in | tee Si.out电子密度可视化,运行以下命令生成XSF文件:
createXSF Si.out Si.xsf n可以用VESTA打开Si.xsf:
进阶
布里渊区采样对能量的影响
修改Si.in中的k点折叠命令:
kpoint-folding ${nk} ${nk} ${nk}编写批量计算脚本run.sh:
#!/bin/bash
for nk in 1 2 4 8 12 16; do
export nk # 将nk设为环境变量
mpirun -n 4 jdftx -i Si.in | tee Si-$nk.out
done
listEnergy Si-?.out Si-??.out运行后得到能量收敛结果:
-7.317807660097655 Si-1.out
-7.849193634824133 Si-2.out
-7.936090648821331 Si-4.out
-7.942846959204424 Si-8.out
-7.942984013789327 Si-12.out
-7.942989279183072 Si-16.out当k点密度达到8x8x8时,能量已收敛至0.0001 Hartree以内。
Monkhorst-Pack网格对比
在Si.in中添加:
kpoint 0.5 0.5 0.5 1.重新运行脚本,得到结果:
-7.849461799239376 Si-1.out
-7.937020474685965 Si-2.out
-7.942892147278267 Si-4.out
-7.942989527101191 Si-8.out
-7.942989655201798 Si-12.out
-7.942989659123112 Si-16.outMonkhorst-Pack网格收敛更快,但对称性简化的k点数(nStates)更多,计算时间更长。
溶液中的分子计算:溶剂化模型入门
化学反应常发生于溶液中,而利用DFT预测反应机理需计算溶液中分子的结构和自由能。直接通过溶剂分子进行全原子描述需数千次DFT计算以采样所有溶剂构型,计算量极大。实际可行的替代方案是使用连续介质溶剂化模型,直接近似溶剂对溶质的平衡效应,而无需处理单个溶剂分子的构型。
JDFTx的核心开发方向之一,是在联合密度泛函理论(Joint Density Functional Theory,JDFT)框架下构建从连续介质到经典DFT的多层次溶剂化模型(JDFTx的"J"即来源于此)。本教程演示如何将分子置于溶液中,并介绍控制溶剂化和流体性质的相关命令。
这里介绍一下 include 命令,这个命令就相当于 latex 的 \input 命令,include 命令可以把一个文件的内容包含到当前文件中。
借用此命令,我们可以把一些公共的设置放在一个文件中,然后在其他文件中引用它们。这样可以避免重复编写相同的代码。也可以把冗长的结构文件单独放在一个文件中,然后在主输入文件中引用它们。
以水分子溶于液态水为例。该过程的自由能与液态水的蒸气压相关,实验测量值为 -0.0101 Hartrees(约 -6.33 kcal/mol)。
首先进行真空中的计算:
公共输入文件 common.in:
# 保存为 common.in
lattice Cubic 15 # 15玻尔半径的立方晶格
coulomb-interaction Isolated # 孤立体系库仑作用
coulomb-truncation-embed 0 0 0 # 截断嵌入位置
ion-species GBRV/$ID_pbe.uspp # 使用GBRV PBE赝势
elec-cutoff 20 100 # 电子截止能设置
coords-type cartesian # 笛卡尔坐标真空计算输入文件 Vacuum.in:
include common.in # 相当于把 common.in 复制到这里
ion O 0.00 0.00 0.00 0 # 氧原子坐标 (0,0,0)
ion H 0.00 1.13 +1.45 1 # 氢原子1坐标 (0,1.13,1.45)
ion H 0.00 1.13 -1.45 1 # 氢原子2坐标 (0,1.13,-1.45)
ionic-minimize nIterations 10 # 离子位置优化(10步)
dump-name Vacuum.$VAR # 输出文件名
dump End State # 输出最终态保存为 Vacuum.in,运行:jdftx -i Vacuum.in | tee Vacuum.out 此计算将输出真空中水分子的优化几何结构和波函数。
溶液计算(LinearPCM 模型) 基于真空计算结果进行溶液化计算:
LinearPCM输入文件 LinearPCM.in:
include common.in
include Vacuum.ionpos # 导入真空优化后的离子位置
initial-state Vacuum.$VAR # 初始态为真空计算结果
ionic-minimize nIterations 10 # 离子位置优化(10步)
dump-name LinearPCM.$VAR # 输出文件名
dump End State BoundCharge # 输出最终态及束缚电荷密度
fluid LinearPCM # 线性响应连续介质流体模型
pcm-variant GLSSA13 # 默认腔体定义方法(同VASPsol)
fluid-solvent H2O # 溶剂为水保存为 LinearPCM.in,运行:jdftx -i LinearPCM.in | tee LinearPCM.out
# Energy components:
A_diel = -0.0148096510994770
Eewald = 3.7898040778182858
EH = 18.3419817002986534
Eloc = -45.6866747367599331
Enl = 2.2303181577355398
Exc = -4.3464926181683348
Exc_core = 0.0650489777314031
KE = 8.3412775388691465
-------------------------------------
Etot = -17.2795465535747148输出文件中:
Linear fluid 段落表示流体自由度优化(通过线性Poisson-Boltzmann方程求解)。
能量项新增 Adiel,表示溶剂的贡献。
溶剂化自由能 = LinearPCM.out 最终能量 - Vacuum.out 最终能量。
本例结果为 -0.0113 Hartrees,与实验值 -0.0101 Hartrees 吻合。
通过以下命令可视化溶剂中的束缚电荷密度:
createXSF LinearPCM.out LinearPCM.xsf nbound相比于VASPSol,JDFTx 提供了更多的溶剂化模型包括:
- LinearPCM
- NonlinearPCM
- SaLSA
- ClassicalDFT
其中LinearPCM或NonlinearPCM又有很多variant,以确定空腔和相关能量(cavitation, dispersion等)。
变体包括:
- CANDLE
- SCCS
- GLSSA13 (VASPsol)
- ...
常用溶剂化模型对比
| 模型 | 特点 | 命令示例 |
|---|---|---|
| LinearPCM (GLSSA13) | 线性响应,腔体由溶质电子密度定义,广泛用于固态计算(如VASPsol) | fluid LinearPCM / pcm-variant GLSSA13 |
| SCCS | 自洽连续介质溶剂化模型(Quantum Espresso实现),腔体定义方法与参数不同 | fluid LinearPCM / pcm-variant SCCS_* |
| CANDLE | 最新推荐模型,结合非局域腔体定义与局部介电响应,修正电荷不对称性 | fluid LinearPCM / pcm-variant CANDLE |
| NonlinearPCM | 非线性介电/离子响应,适用于电化学电容计算 | fluid NonlinearPCM / pcm-variant GLSSA13 |
| SaLSA | 非局域角动量展开响应,参数少但计算耗时(约10倍于线性模型) | fluid SaLSA |
| ClassicalDFT | 全原子经典DFT描述溶剂微观结构,计算耗时极高(约100倍于线性模型) | fluid ClassicalDFT |
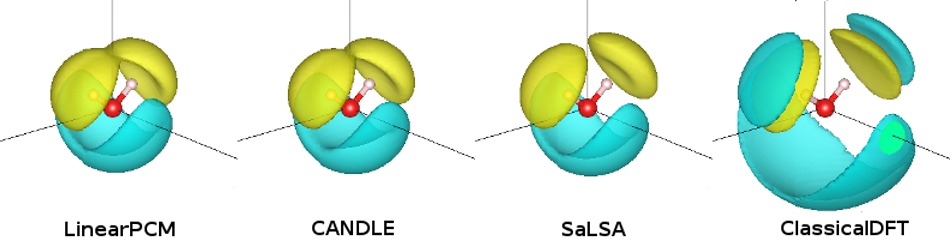
电荷可视化:通过 createXSF 生成各模型的 nbound 电荷密度,观察:
LinearPCM、CANDLE、SaLSA 的电荷集中在溶质表面。
ClassicalDFT 显示多层壳状溶剂结构。
带电表面与电化学系统计算
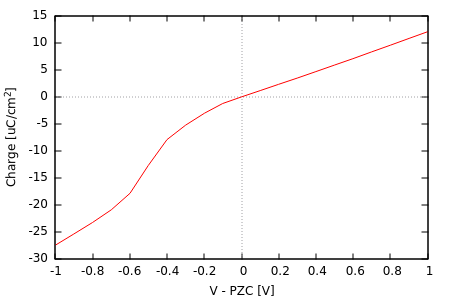
结合电解质(溶剂+离子)的连续介质溶剂化模型,我们可以直接处理溶液中的带电表面,这对电化学系统尤为重要。电极表面通过外部电路设定电子化学势(mu),电子数量随mu平衡,导致表面带电(除零电荷电位PZC外)。
本教程以Pt(111)表面的电荷-电势曲线计算为例,演示恒电势计算(通过target-mu命令直接设定电子化学势)。
以下是关于 target-mu 命令的详细解释:
target-mu <mu> [<outerLoop>=no]
<mu>:设定电子化学势的绝对值(以Hartree为单位,相对于真空能级)。
此命令用于恒电势计算(固定化学势mu,而非固定电荷数),模拟电化学系统中电极与外部电路接触时的电子交换行为。
化学势 <mu> 的设定
mu是绝对量,需根据参考电极校准。例如:
相对于标准氢电极(SHE)的电势V(单位:伏特)转换为:
mu = -(Vref + V)/27.2114Vref:SHE相对于真空能级的电位(实验值约4.44 V)。
27.2114:Hartree与eV的转换系数(1 Hartree 约 27.2114 eV)。
示例:若需模拟相对于SHE为+0.5 V的电位:
target-mu -(4.44 + 0.5)/27.2114物理背景
巨自由能(G):
恒电势下系统的热力学势为 G = F - muN,其中 F 是亥姆霍兹自由能,N 是电子数。此量在固定mu时具有变分最小性。
与固定电荷计算的差异:固定电荷时优化 F,而固定mu时优化 G,更贴近真实电化学界面条件。
设置target-mu后将会直接进行巨正则系综变分优化:mu全程固定,电子数连续调整。
输出文件中的自由能标记为 G(巨自由能),而非固定电荷时的 F。
电子数 N 会在迭代中动态更新,直至与mu平衡。
公共输入文件 common.in:
# 保存为 common.in
lattice Hexagonal 5.23966 36 # 六方晶格,基矢长5.23966玻尔,高度36玻尔
ion Pt 0.333333 -0.333333 -0.237676 1 # Pt原子位置(分数坐标)
ion Pt -0.333333 0.333333 -0.118838 1
ion Pt 0.000000 0.000000 0.000000 1
ion Pt 0.333333 -0.333333 0.118838 1
ion Pt -0.333333 0.333333 0.237676 1
ion-species GBRV/$ID_pbesol.uspp # 使用PBEsol赝势
elec-ex-corr gga-PBEsol # 交换关联泛函
elec-cutoff 20 100 # 电子截止能设置
coulomb-interaction Slab 001 # 平板库仑作用(沿z方向)
coulomb-truncation-embed 0 0 0 # 截断嵌入位置
kpoint-folding 12 12 1 # k点网格12x12x1
elec-smearing Fermi 0.01 # 费米展宽0.01 Hartree
fluid LinearPCM # 线性PCM溶剂模型
pcm-variant CANDLE # CANDLE腔体定义
fluid-solvent H2O # 溶剂为水
fluid-cation Na+ 1. # 阳离子为Na+(浓度1M)
fluid-anion F- 1. # 阴离子为F-(浓度1M)
dump-name common.$VAR # 输出文件名(后续计算覆盖)
initial-state common.$VAR # 初始态为前次计算结果
dump End State BoundCharge # 输出最终态及束缚电荷中性表面计算
输入文件 Neutral.in:
include common.in
electronic-SCF # 电子自洽场计算运行:
jdftx -i Neutral.in | tee Neutral.out中性计算结果为后续带电表面计算提供初始状态和PZC(零电荷电位)。
恒电势计算
输入文件 Charged.in:
include common.in
electronic-minimize nIterations 200 # 电子最小化(200步)
target-mu ${mu} # 设定电子化学势mu使用Bash脚本批量运行不同mu值计算:
#!/bin/bash
for iMu in {-10..10}; do
# 计算当前mu(相对于PZC -0.2015 Hartrees,步长0.1 eV)
export mu="$(echo $iMu | awk '{printf("%.4f", -0.2015 + 0.1*$1/27.2114)}')"
mpirun -n 4 jdftx -i Charged.in | tee Charged$mu.out
mv common.nbound Charged$mu.nbound # 重命名束缚电荷文件
done保存为run.sh,运行:
chmod +x run.sh
./run.sh脚本将mu从-1 eV到+1 eV(相对于PZC)分21步计算,每次调整0.1 eV。首次计算耗时较长,后续计算因继承前次状态加速。
# Energy components:
A_diel = -0.0595853544702656
Eewald = 4702.9680318706014077
EH = 5113.6626774397536792
Eloc = -10338.4976578717942175
Enl = -5.2756435006285551
Exc = -141.6058874747504888
Exc_core = 81.2082896017071789
KE = 142.4800516625221860
MuShift = -0.0081434898361512
-------------------------------------
Etot = -445.1278671168959136
TS = 0.0560709862761570
-------------------------------------
F = -445.1839381031720677
muN = -14.0948751579097209
-------------------------------------
G = -431.0890629452623557这里是计算后的一个输出文件,其中G就是自由能。
数据分析与可视化
- 提取电荷-电势数据:
#!/bin/bash
for file in Charged*.out; do
awk '/FillingsUpdate/ {mu=$3; N=$5} END {print mu, N}' "$file"
done保存为collectResults.sh,运行:
chmod +x collectResults.sh
./collectResults.sh | tee mu_N.dat- 使用gnuplot绘制电荷密度曲线:
A = 2*((5.23966*0.529e-8)**2)*sin(pi/3) # 表面积(cm2,上下表面)
qe = -1.602e-13 # 电子电荷(uC)
mu0 = -0.2015 # PZC对应mu
N0 = 80 # 中性表面电子数
set xlabel "V - PZC [V]"
set ylabel "Charge [uC/cm2]"
set xzeroaxis
set yzeroaxis
unset key
plot "mu_N.dat" u ((mu0-$1)*27.2114):(($2-N0)*qe/A) w l结果将显示Pt(111)表面电荷随电势变化曲线,正负区斜率差异反映CANDLE模型的电荷不对称性。
本文主要讲述了 JDFTx 基本的用法、溶剂化和固定电势模拟,相比于 VASP,JDFTx 有以下优势:
- 提供变量支持,提供更灵活的操作
- 支持 GPU 加速,加速效率很客观
- 提供更多的隐式溶剂化模型
- 内置了恒电势算法,通过
target-mu命令指定电势后,JDFTx 将会固定mu自动优化电子数到指定值。
缺点:
- 计算速度比VASP慢
最后,本文内容全部来自于官网,官网还提供了非常详细的例子和文档,感兴趣的同学可以去看看。